
· are interested in genetic testing for autism
·
are affected by miss-expression of the
genes:-
·
ANKRD17
·
TCF4
·
CNTNAP2
·
NRXN1
It is one of those posts that could go on forever; the more you dig, the more you uncover and you wonder why other people (salaried researchers) are not doing this.
Today’s post is mainly about a gene called ANKRD17, but it does highlight more general
principles about those genetic testing results, some parents strive to
obtain. It does look at downstream
effects of Pitt Hopkins Syndrome and “Pitt Hopkins-like Syndrome”, which likely merge into mainstream autism.
Many single gene autisms have already been identified, some have names and some are still SWANs (Syndromes Without A Name). Some syndromes have long been identified, but their biological basis had not been identified. From last year, loss of function of the gene ANKRD17 became Chopra-Amiel-Gordon Syndrome.
Our reader Mary’s whole exome sequencing (WEG) from a few
years ago, for her daughter, has now been flagged up as carrying a mutation
leading to Chopra-Amiel-Gordon Syndrome.
In effect, the mutation in ANKRD17 went from be of no confirmed relevance to autism,
to being causal, thanks to Dr Chopra and her pals.
This highlights the weakness in the
interpretation of genetic testing. Any
benchmark list of autism genes is just a work in progress; your mutation may
not yet be there.
Another gene recently queried by a
reader was CNTNAP2,
this turns out to a key DEG (differentially expressed gene) of a syndrome with
its own name, Pitt Hopkins Syndrome, caused by reduced expression of TCF4
(Transcription Factor 4). Reduced
expression of TCF4 has very many effects, but one effect is to reduce expression of
CNTNAP2.
In lay-speak, lack of TCF4 causes a cascade of effects, one of which is on the expression of CNTAP2. We see that people with a CNTAP2 mutation share many of the features of people having a TCF4 mutation. So, of all the many effects caused by TCF4, those along the TCF4-CNTAP2 pathway should be focused on. The mutation in CNTNAP2, quite rationally, is now called Pitt Hopkin-like Syndrome-1. There is also a Pitt Hopkin-like Syndrome-2 which is caused by a mutation in NRXN1 (neurexin 1).
https://royalsocietypublishing.org/doi/pdf/10.1098/rsob.210091
In mammals, the neurexins are
encoded by three NRXN genes (NRXN1-3), each of which has both an upstream
promoter that is used to generate the α-neurexins, and a downstream promoter
that is used to generate the shorter β-neurexins [13,15].
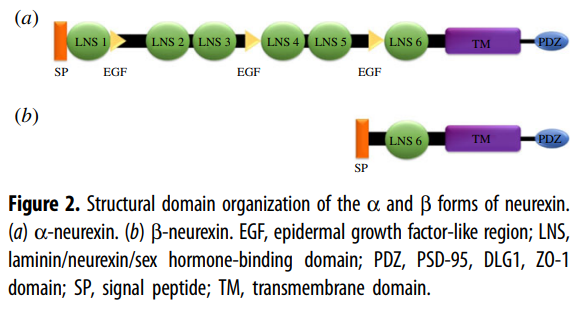
α-neurexins are composed of six large extracellular
laminin/neurexin/sex hormone-binding (LNS) globulin domains with three
interspersed epidermal growth factor (EGF)-like regions
Just note the term EGF.
In very recent research we see that a reduction in epithelial growth factor may be
what is driving some of the key clinical features, such as lack of language.
CNTNAP2 has been identified as a master gene in autism manifestation responsible for speech-language delay by impairing the EGF protein domain and downstream cascade. The decrease in EGF is correlated with vital autism symptoms, especially language disabilities.
Autism exhibits genetic heterogeneity, and
hence, it becomes difficult to pinpoint one single gene for its manifestation.
The gene clusters with varied pathways show the convergence of multiple gene
variants, resulting in autism manifestation. Whole-exome sequencing proves to
be a reliable tool for deciphering the causal genes for autism manifestation.
Deciphering the autism exome identified the mutational landscape derived from
single and multi-base DNA variants. Genes carrying mutations were identified in
synaptogenesis processes, EGF signaling, and PI3K/MAPK signaling. Protein-protein interactions of
NrCAM and CNTN4 with CNTNAP2 increased the impact and burden on autism.
Shining a light on CNTNAP2: complex
functions to complex disorders
TCF4 encodes a basic helix-loop-helix (bHLH) transcription factor that binds near the start site of CNTNAP2 to upregulate its expression (Figure 1a).48 In humans, TCF4 is more highly expressed in the neocortex and hippocampus than in the striatum, thalamus and cerebellum.49 Mutations in TCF4 have been shown to cause Pitt–Hopkins syndrome (PTHS) and three rare TCF4 SNPs are associated with schizophrenia.17, 49, 50, 51 PTHS is characterised by severe intellectual disability, absent or severely impaired speech, characteristic facial features and epilepsy.52 Many of these features are shared with patients carrying CNTNAP2 mutations, leading researchers to test patients with PTHS-like features for CNTNAP2 mutations.17 Two mutations affecting the CNTNAP2 locus (one homozygous and one compound heterozygote) were identified in two independent pedigrees (Table 1). This suggested that disruption of the TCF4–CNTNAP2 pathway could be related to intellectual disability, seizures, and/or behavioural abnormalities.
One of our readers in Australia recently queried the potential
significance of a mutation (an SNP) in CNTNAP2.
Based on the above, it clearly could be very important.
What is the common link between TCF4, CNTNAP2 and NRXN1? It would
seem to be EGF (epidermal growth
factor).
It looks quite possible that EGF is
disturbed in much broader autism. It appears that inflammation may reduce EGF
levels. It is a rather circular argument, but we also know that EGF reduces
inflammation.
To sum up people, with autism likely want more EGF and we already knew that they definitely want less inflammation.
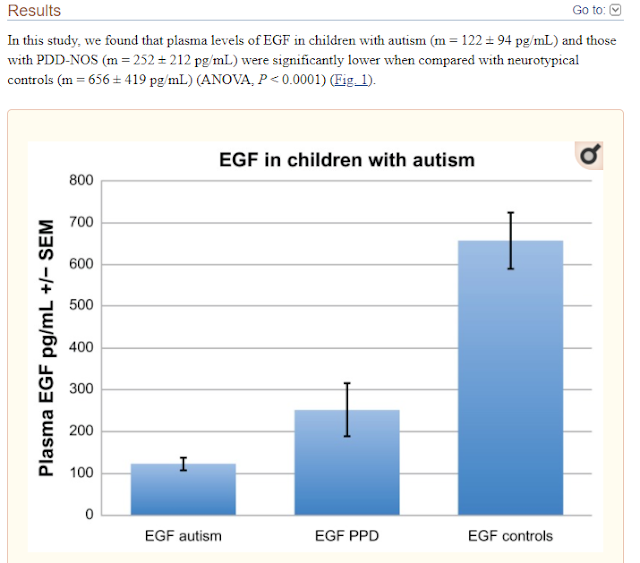
Decreased Epidermal Growth Factor (EGF) Associated with HMGB1 and Increased Hyperactivity in Children with Autism
These results suggest an association between
decreased plasma EGF levels and selected symptom severity. We also found a
strong correlation between plasma EGF and HMGB1, suggesting inflammation is
associated with decreased EGF.
ANKRD17
Finally, we get back to ANKRD17.
Our reader Mary has already highlighted
this recent paper: -
Heterozygous ANKRD17 loss-of-function variants cause a
syndrome with intellectual disability, speech delay, and dysmorphism

Dysmorphic facial features of the ANKRD17-related
disorder
ANKRD17 is an ankyrin repeat-containing
protein thought to play a role in cell cycle progression, whose ortholog
in Drosophila functions
in the Hippo pathway as a co-factor of Yorkie. Here, we delineate a
neurodevelopmental disorder caused by de
novo heterozygous ANKRD17 variants.
The mutational spectrum of this cohort of 34 individuals from 32 families is
highly suggestive of haploinsufficiency as the underlying mechanism
of disease, with 21 truncating or essential splice site variants, 9 missense
variants, 1 in-frame insertion-deletion, and 1 microdeletion (1.16 Mb).
Consequently, our data indicate that loss of ANKRD17 is
likely the main cause of phenotypes previously associated with large
multi-gene chromosomal aberrations of the 4q13.3 region. Protein
modeling suggests that most of the missense variants disrupt the stability
of the ankyrin repeats through alteration of core structural residues. The major phenotypic
characteristic of our cohort is a variable degree of developmental
delay/intellectual disability, particularly affecting speech, while additional
features include growth failure, feeding difficulties, non-specific MRI
abnormalities, epilepsy and/or abnormal EEG, predisposition to recurrent
infections (mostly bacterial), ophthalmological abnormalities, gait/balance
disturbance, and joint hypermobility. Moreover, many individuals shared similar
dysmorphic facial features. Analysis of single-cell RNA-seq data from
the developing human telencephalon indicated ANKRD17 expression
at multiple stages of neurogenesis, adding further evidence to the
assertion that damaging ANKRD17 variants cause a neurodevelopmental
disorder.
Neonatal growth parameters were normal in the majority of
individuals (Table S2) but postnatal
growth failure was a feature of almost half of the individuals (height
< 2 SD in n ¼ 12 and weight < 2 SD in n ¼ 9). One individual with marked
growth failure (individual 3, height 3.8 SD) was under treatment with growth
hormone (GH), although GH stimulation testing was normal. Feeding difficulties,
especially reduced oral intake, were reported at some stage in 11 individuals,
5 of whom required G-tube nutritional supplementation. Postnatal microcephaly (OFC < 2SD) was noted
in seven individuals, and macrocephaly in four (one of these individuals, however, also
harbored a pathogenic de novo NSD1 variant (GenBank: NM_022455.4, c.2615T>G
[p.Leu872*]). Epilepsy was
reported in nine individuals (individuals 1, 2, 16, 19, 21, 25, 27, 28,
and 33), with an age of onset of under 2 years for five individuals
(individuals 1, 2, 16, 19, and 25). Focal seizures with secondary
generalization was the most common seizure subtype, present in five individuals
(individuals 1, 2, 21, 25, and 27). One individual had Lennox-Gastaut epilepsy
(individual 16), one had tonic seizures with head deviation (individual 19),
one had mixed myoclonic and tonic-clonic epilepsy (individual 33), and another
a mixture of tonic-clonic and absence seizures (individual 28). Seizures were
well controlled (less frequent than every 2 years) in five individuals (individuals
2, 21, 25, 28, and 33), all of whom were on three or fewer antiepileptic drugs
(AEDs). Moderate control, with seizures every 2–3 months, was reported in
individual 1, who was on Valproate monotherapy. Two individuals had refractory
epilepsy during at least parts of their disease course—individual 19 who had
frequent tonic seizures in infancy that resolved with topiramate monotherapy
and individual 16 Table 2. Frequencies of phenotypic characteristics of
individuals with ANKRD17 variants Frequency Sex F ¼ 19, M ¼ 15 Growth Height
< 2 SD 12/31 Weight < 2 SD 9/30 OFC < 2 SD 7/31 OFC > 2 SD 4/31
Development DD or ID 31/34 severe 7 moderate 12 mild 5 borderline 7 Motor delay
20/29 Speech delaya 29/32 Other ASD, n ¼ 8; ADHD, n ¼ 4 Neurology Epilepsy 9/33
Abnormal EEG 10/23 Brain MRI abnormalities 11/23 Gait or balance abnormalities
9/25 Spasticity or hypertonia 4/26 Other Recurrent infections 11/33 Feeding problems 11/27 Palate
abnormalities 3/34 Hypermobility 9/29 Ophthalmological abnormalities 13/23 Miscellaneous
Minor digital anomalies 6 Genitourinary abnormalities 5 Pigmentary
abnormalities 4 Scoliosis 3 Abnormal bone mineralization 2 Prominent blood
vessels 2 ADHD, attention deficit hyperactivity disorder; ASD, autism spectrum
disorder a For details see Table S1 The American Journal of Human Genetics 108,
1138–1150, June 3, 2021 1143 who had multiple seizures every day despite three
AEDs. Further details of epilepsy phenotype, including previously trialled
AEDs, are noted in Table S2. There were four individuals without epilepsy in
whom an abnormal EEG was recorded. Other neurological features include poor
balance and/or abnormal gait (9/25) and peripheral spasticity (4/26, one of
whom one was microcephalic). Neuroimaging
abnormalities were identified in 11 of the 23 individuals in whom an MRI was
recorded. Abnormalities include decreased white matter volume (individuals 14,
16, and 18), thinning of the corpus callosum (individuals 14 and 19), optic
nerve hypoplasia (individuals 18 and 19), a localized hyperintensity
(individuals 7 and 31), right temporal sclerosis (individual 16), dilated
Virchow-Robin spaces (individual 6), periventricular nodular heterotopia
(individual 30), and an arachnoid (individual 24) and pineal cyst (individual
16). Ophthalmological
abnormalities were reported in 13/23 individuals. There were nine individuals with
recurrent bacterial infections, one with recurrent viral infections, and
one individual with recurrent infections that were both viral and bacterial.
The source of bacterial infection was primarily the upper and lower respiratory
system and the middle ear (nine individuals) and in some cases required
hospitalization. Two individuals were on low-dose prophylactic antibiotics for
recurrent otitis media or respiratory tract infections. Notably, individual 26
had a history of pseudomonas and methicillin-resistant staphylococcal aureus
(MRSA) infection on his toes. Immunology assessments were recorded in five
individuals, details of which can be found in Table S2, with no obvious
immunodeficiency identified in these individuals. Generalized joint
hypermobility was reported in 9/29 individuals. Notably, there were two
individuals with cleft palate in the context of Pierre Robin sequence (PRS) and
another with cleft lip and palate. Other infrequent features include minor
digital anomalies (n ¼ 6), genitourinary abnormalities (n ¼ 5, of whom three
had unilateral renal agenesis), abnormal skin pigmentation (n ¼ 4), scoliosis
(n ¼ 3), abnormality of bone mineralization (n ¼ 2), and cutaneous prominence
of blood vessels (n ¼ 2). Figure 2 shows the facial features of individuals
with the ANKRD17-related neurodevelopmental disorder. Key dysmorphic features
include a triangular-shaped face found in 10 of the 24 individuals for whom photos
were available with a high anterior hairline (19/24), eyes which are either
deep-set (5/24) or almond shaped (8/24) with periorbital fullness (6/24), thick
nasal alae and flared nostrils (9/24), full cheeks (7/24), and a thin upper lip
(12/24). The degree of dysmorphism was variable, with several individuals
(particularly individuals 8 and 10) presenting with only subtle dysmorphic
characteristics. Persistence of the high anterior hairline, periorbital
fullness, and full cheeks into adulthood is demonstrated in individual 12 (age
30 years) and individual 25 (age 34 years). A number of diagnoses had been
considered in several individuals prior to the identification of an ANKRD17
variant, including SATB2-associated syndrome (MIM: 612313) in individual 5 who
presented with PRS, triangular facies and speech delay, and Floating-Harbour
syndrome (MIM: 136140) in individual 9 who presented with marked short stature
(height < 3 SD), microcephaly (head circumference < 2.5 SD), dysmorphic
features, and borderline ID. This
highlights the phenotypic overlap of the ANKRD17-related disorder with a number
of other genetic syndromes, notably those with expressive language delay. In
our cohort, significant speech delay was reported in most individuals (n ¼ 29)
even in those with IQ in the borderline range. The finding that verbal IQ was
reduced relative to performance IQ in three of the five individuals for whom
deep neuropsychological phenotyping was available adds further evidence to our
observation that expressive language is particularly affected in this disorder
How were the 34
individuals identified?
In the Table 1 of the paper, we see
that the great majority of the children had been identified from WES (whole
exome sequencing), a few had WGS (whole genome sequencing) and just one via
micro array testing.
They families clearly opted to share
their data, in the hope of some researcher finding it useful later, as Chopra, Amiel and
Gordon clearly did after a few years later.
How do you figure
out the DEGs (differentially expressed genes)?
To treat ANKRD17 deficiency (now known as Chopra Amiel Gordon Syndrome) you have a choice.
·
Increase expression of ANKRD17 via gene therapy, or a drug (if that
were possible)
·
Treat some of the
downstream DEGs (Differentially Expressed Genes)
Mary asked how you could identify the
DEGs, given there is only one paper published on Chopra Amiel Gordon Syndrome.
You can start by reviewing everything known about ANKRD17.
A very good place to start is on the
GeneCards website.
https://www.genecards.org/cgi-bin/carddisp.pl?gene=ANKRD17
Most people will
end up having to learn some new words to understand everything on the above website.
The first thing to note is just how
wide ranging are the functions of this gene and this accounts from the wide-ranging
problems associated with it. It even plays
a role in dealing with both viral and bacterial infections.
It is particularly upregulated in the
fetal brain and that likely leads to the autism/ID related effects.
Protein
differential expression in normal tissues from HIPED for ANKRD17 Gene
This gene is overexpressed in Lung (18.9), Platelet (15.4),
Retina (8.4), and Fetal Brain (6.4).
We can see that this gene is
associated with Chopra Amiel Gordon Syndrome and
Non-Specific Syndromic Intellectual Disability.
Quite possibly, Non-Specific Syndromic Intellectual
Disability was used as a term because Chopra Amiel Gordon Syndrome did
not yet exist.
But is useful to look up Non-Specific Syndromic Intellectual
Disability, to see which other genes are listed. This then tells you much about what can cause
ID. Follow the link below.
https://www.malacards.org/card/non_specific_syndromic_intellectual_disability
We see a very long list of syndromes and genes.
There are 61 genes listed.
Going back to the Genecards ANKRD17 page, we can see
if there are known protein interactions that might result in autism/ID.

EIF4E2 does look familiar, and I recall a link to Fragile X. So, I looked it up.
Note that we see both EIF4E and EIF4E2 - Eukaryotic Translation Initiation Factor 4E Family Member 2. Note that is has a second name, 4EHP.
EIF4E2 is a version/homolog of EIF4E
EIF4E2 = 4EHP
The eIF4E homolog
4EHP (eIF4E2) regulates hippocampal long-term depression and impacts social behavior
Background: The regulation of protein synthesis is a critical
step in gene expression, and its dysfunction is implicated in autism spectrum
disorder (ASD). The eIF4E homologous protein (4EHP, also termed eIF4E2) binds
to the mRNA 5' cap to repress translation. The stability of 4EHP is maintained through
physical interaction with GRB10 interacting GYF protein 2 (GIGYF2).
Gene-disruptive mutations in GIGYF2 are linked to ASD, but causality is
lacking. We hypothesized
that GIGYF2 mutations cause ASD by disrupting 4EHP function.
4EHP is
expressed in excitatory neurons and synaptosomes, and its amount increases
during development. 4EHP-eKO mice display exaggerated mGluR-LTD, a phenotype
frequently observed in mouse models of ASD.
Conclusions: Together these
results demonstrate an important role of 4EHP in regulating hippocampal
plasticity and ASD-associated social behaviors, consistent with the link
between mutations in GIGYF2 and ASD.
The disruption of protein
synthesis (mRNA translation or translation) in the brain by genetic
perturbations of its regulators constitutes a known underlying etiology for ASD
[2, 3]. For
most mRNAs, initiation of translation requires binding of initiation factors to
their 5′ end at a modified guanine nucleotide (m7GpppN, where N is
any nucleotide) termed the 5′ cap [4]. The
eukaryotic initiation factor (eIF) 4F complex is comprised of the cap binding
protein eIF4E, an mRNA helicase eIF4A, and a molecular scaffold eIF4G. Together
these proteins facilitate recruitment of the ribosomal 43S preinitiation
complex to the mRNA. Overactivity
of eIF4E in humans has been implicated in ASD [5, 6] and ASD-like phenotypes in mice [7, 8]. Indeed, disruption of the proteins regulating eIF4E
activity, such as fragile
X mental retardation protein (FMRP)
[9], cytoplasmic FMR1 interacting protein 1 (CYFIP1) [10], and eIF4E-binding protein 2 (4E-BP2) [8, 11, 12], is implicated in ASD. It is therefore necessary to
investigate the function of ASD-linked genes that encode for regulators of translation.
Whole-genome sequencing of ASD patients has been invaluable in identifying
these genes.
If you look up the protein interaction for the Fragile X gene (FMRI), you do indeed see EIF4E close by. FMR1 encodes the fragile X mental retardation protein.
This blog is full of ideas regarding treating Fragile X,
because there are so many studies of that type of autism.
It is rather mind-boggling that there are still no approved
therapies for Fragile X. The same holds
true for Down Syndrome (DS). This is a
recurring story, where it pays to be the early adopter, not one of the passive
followers.
Leaky
ATP from either Mitochondria or Neurons in Fragile X and Autism
In that post I suggested “Mirapex - a
miracle for Fragile X?”
In the post below we saw how EIF4E leads to autism, and how
FMRP from Fragile-X affects EIF4E.
Vasopressin,
Oxytocin, the Lateral Septum, Aggression and Social Bonding, Autism gene NLGN3
and MNK inhibitors for reversing Fragile-X and likely more Autism

One of the papers below goes further
and suggests
“This
work uncovers an unexpected convergence between the genetic autism risk factor
Nlgn3, translational regulation, oxytocinergic signalling, and social novelty
responses”
“We
propose that pharmacological inhibition of MNKs may provide a new therapeutic
strategy for neurodevelopmental conditions with altered translation
homeostasis”
“Our
work not only highlights a new class of highly-specific, brain-penetrant MNK
inhibitors but also expands their application from fragile X syndrome to a
non-syndromic model of ASD”
Regarding
Fragile X
“Collectively, this work establishes eFT508 (an MNK inhibitor) as a
potential means to reverse deficits associated with FXS.”
Conclusion
My quick
look at the subject suggests that, amongst other likely DEGs, the NLGN (neuroligin)
genes are quite possibly miss-expressed.
In
humans, alterations in genes encoding neuroligins are implicated in autism and other cognitive disorders.
In
Genecards the association is with EIF4E2 rather than the EIF4E, which we
know affects neuroligin expression. But EIF4E2 is just a version of EIF4E.
These protein interaction maps are not perfect and different
sources often come up with slightly different maps.
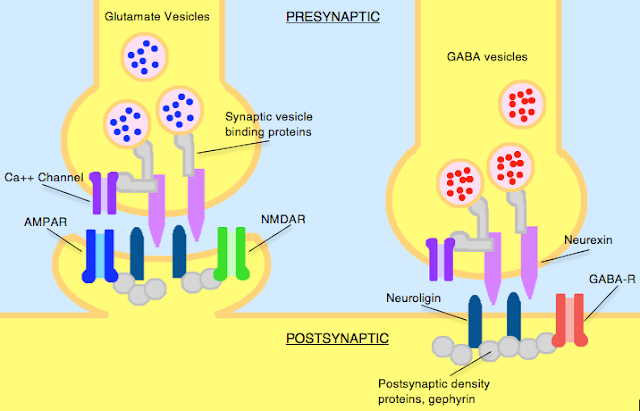
What are Neurexins and Neuroligins?
Neurexins and neuroligins are synaptic cell-adhesion molecules that connect pre- and postsynaptic neurons at synapses, they mediate signalling across the synapse, and shape the properties of neural networks by specifying synaptic functions. Neurexins and neuroligins are therefore very important and can be dysfunctional in autism.
It looks
like growth signaling is disturbed in Chopra-Amiel-Gordon Syndrome, but it not always in the same way. Both too much and too
little growth are possible.
An MRI
would not be a bad idea, and measuring the corpus callosum would be helpful.
The corpus callosum connects the right and left side of the brain and is the largest
white matter structure in the brain, which means lots of myelin should be
there.
If it is very narrow, that would tell you something, hopefully it is normal. You cannot really change its size, but if it lacked myelination that might be something you could affect.
Positive Correlations
between Corpus Callosum Thickness and Intelligence
Trying the cheap and partially effective treatments for fragile X might be helpful. It is possible that the Fragile X DEGs overlap with the Chopra-Amiel-Gordon Syndrome DEGs.
The
following drugs are cheap generics that are helpful, to some extent, in
Fragile-X.
·
Metformin
·
Lovastatin
·
Baclofen
As the altered E/I balance is present in Fragile X and most autism, it would be worthwhile trying the E/I corrective therapies that exist, in case one is beneficial. There are different causes of an E/I imbalance, but since there are not many therapies, it is easier to just try them one by one.
It is
also highly likely that common features of autism may be present, such as
·
oxidative stress (NAC)
·
neuroinflammation (numerous therapies)
·
impaired myelination (Clemastine, Ibudilast, NAG) NAG
is not the same as NAC, it is N-acetylglucosamine
·
mitochondrial dysfunction (Carnitine, antioxidants, activate
PGC-1 alpha via PPAR gamma e.g. with Pioglitazone)
·
folate receptor antibodies (Calcium folinate)
If the
Corpus Callosum is smaller than it should be, or is demyelinated, you could try
high bio-availability curcumin, in addition to the above pro-myelination
therapies.
Which ion
channel dysfunctions appear in Chopra-Amiel-Gordon Syndrome? I did not see any
clues, but where there is epilepsy, there is very likely going to be an ion
channel dysfunction involved.
My son has a gene mutation on DDX3X which has been associated with intellectual disability and reduced corpus callosum size. What treatments have been shown to assist with this issue?
ReplyDelete-TMM
TMM, conventional wisdom is that you can do nothing.
DeleteBut, if you look up your gene, you will see that among the top 5 interactions is the EIF4E gene from todays post. The one affected in Fragile X. So what helps in Fragile X is well worth trying.
Look up curcumin and corpus callosum. In mice modified curcumin has some effect. High bioavailable curcumin is available. One reader whose don has agenesis of the corpus callosum found it had some benefit.
Peter,
DeleteThank you for the response. Given that there has not been much research into DDX3X (only discovered in 2014), it has been tough to find therapies that have had any effect. We have tried Memantine, Galantamine, Leucovorin, and Bumetanide with minimal success.
Some of the research that is out there points to a dysregulation in Rac1 and also the Wnt signaling pathway. Here are some examples:
-DDX3 Modulates Neurite Development via Translationally Activating an RNA Regulon Involved in Rac1 Activation (https://pubmed.ncbi.nlm.nih.gov/27656019/):
“Moreover, DDX3 activates the translation of functionally coherent mRNAs involved in Rac1 activation including Rac1 Among the DDX3 regulon, Prkaca encodes the catalytic subunit of PKA, a potential activator of Rac1 in neurons…Inhibition of DDX3 activity or expression in neonatal mice impaired dendritic outgrowth and spine formation of hippocampal neurons, echoing neuronal deficits underling DDX3 mutation-associated ID….The novel DDX3 regulon may conduct a spatial and temporal control of Rac1 signaling to regulate neurite development.”
-DDX3 modulates cell adhesion and motility and cancer cell metastasis via Rac1-mediated signaling pathway (https://www.nature.com/articles/onc2014190):
“We also examined the mechanism underlying DDX3-regulated cell migration. DDX3 knockdown reduced the levels of both Rac1 and β-catenin proteins, and consequentially downregulated the expression of several β-catenin target genes.”
-Mutations in DDX3X Are a Common Cause of Unexplained Intellectual Disability with Gender-Specific Effects on Wnt Signaling (https://www.sciencedirect.com/science/article/pii/S0002929715002803#bib35)
“Taken together, our in vivo testing of the nonsynonymous DDX3X mutations demonstrated marked alteration in Wnt-mediated ventralization for changes arising de novo in affected females. Our data also reinforce the notion that disruption of β-catenin signaling during neurodevelopment has profound consequences. Loss of Wnt signaling inhibits neuroblast migration, neural differentiation, and suppression of the development of the forebrain…Our phenotypic read out in zebrafish shows a gender difference in identified variants with a loss-of-function effect of all tested de novo mutations on the Wnt pathway.”
It seems that the objective here would be to increase Rac1 and also activate the Wnt pathway. I was reading through some of your previous posts regarding BHB (https://epiphanyasd.blogspot.com/2018/09/ketones-and-autism-part-5-bhb-histone.html):
“Our further analysis showed that the BHB-induced microglial ramification was mediated by the Rac1-Cdc42 signal, as BHB markedly increased the activity of Rac1 and Cdc42”
Ive also been looking into Huperzine A to activate the Wnt pathway given that we have not had any success in locating a doctor that would give a script for a statin for this:
-Huperzine A Activates Wnt/β-Catenin Signaling and Enhances the Nonamyloidogenic Pathway in an Alzheimer Transgenic Mouse Model (https://www.nature.com/articles/npp2010245):
“Importantly, our results further showed that HupA inhibited GSK3α/β activity, and enhanced the β-catenin level in the transgenic mouse brain and in SH-SY5Y cells overexpressing Swedish mutation APP, suggesting that the neuroprotective effect of HupA is not related simply to its AChE inhibition and antioxidation, but also involves other mechanisms, including targeting of the Wnt/β-catenin signaling pathway in AD brain.”
Also, it seems that curcumin would be an inhibitor of DDX3X, and may not be advantageous in our case:
Delete- A Computational Approach with Biological Evaluation: Combinatorial Treatment of Curcumin and Exemestane Synergistically Regulates DDX3 Expression in Cancer Cell Lines
“The obtained results illuminate the use of curcumin as an alternative DDX3 inhibitor and can serve as a chemical scaffold to design new small molecules”
Given this, I am thinking of trying the following:
-Huperzine A – increase Wnt signaling
-BHB – upregulate Rac1
Does that sound reasonable or am I missing something?
Much Thanks,
-TMM
TMM,in general you will find that pharmaceuticals are far more potent than OTC products and even pharmaceuticals are only going to provide “fine tuning” of the brain.
DeleteYou may also discover that sometimes what works is the exact opposite of what the science might initially imply. So, I would start by trying to have an effect and then narrow it down to have a good effect. In many cases you will see very quickly what works, does nothing, or indeed makes things worse.
I would expect the common drug Simvastatin to have an effect. It affects both Wnt signalling and Rac1. It is the opposite effect on Rac1 that you are looking for. I use the very similar Atorvastatin.
BHB is a very interesting substance with many effects that might be beneficial, it even blocks the blocks the NLRP3 inflammasome.
Science can help, but you will inevitably needs some trial and error.
Peter--thank you so much for this tremendously helpful roadmap to understanding ANKRD17 mutations and Chopra-Ameil-Gordon Syndrome. It's going to take me a long time to start to grasp all that you've included here, but I'm anxious to dig in.
ReplyDeleteSince your last post, we've emailed Maya Chopra as you suggested. She very kindly responded. She and her colleagues have completed the protocol for a study of ANKRD17, and it's currently under review. They also plan to look at downstream expression and treatments in the future, but I'm not sure of the timing for that.
Thank you again for giving us this huge head start. I'll keep you updated on our progress.
~Mary
Hi Peter,
ReplyDeleteMy autistic child is 4.5 years old, and he has CNTNAP4 gene duplication (x4) that means 2 extra copies of this gene. Do you recommend any therapy that is going to be beneficial for him especially with this mutation?
Thank you
Maas
Hi Peter, it is Maas again. What do you think about duplications in general, is it different from deletions phenotypically?
ReplyDeleteThank you
Maas, the best researched duplication/deletion syndrome is chromosome 16p11.2.
DeleteDeletion leads to large heads and so also Chiari “brain hernias” when the brain grows down towards the spine. Duplication leads to the opposite, so small heads (microcephaly) and corpus callosum malformation. But features like poor speech articulation and seizures are found in both duplication and deletion of 16p11.2.
This is quite a common phenomenon, too much and too little of something are both harmful, you need just the right amount of gene expression.
This paper is a little old, but is interesting. It is Dutch, which you may also be.
Connecting the CNTNAP2 Networks with Neurodevelopmental Disorders
https://www.karger.com/Article/FullText/371594
I would suggest you contact the author
Martin Poot, PhD
Department of Medical Genetics, University Medical Center Utrecht
Mail Stop: Str. 1.305, PO Box 85090
NL-3508 AB Utrecht (The Netherlands)
E-Mail Martin_Poot@hotmail.com
I think he has moved, but you can track him down.
Potassium channels play a role in CNTNAP2 function. You can block them, open them and indeed change the level of potassium within cells.
Our researcher reader Knut’s therapy for autism changes the level of potassium inside neurons using an OTC pain reliever (Mefenamic Acid / Ponstan). This is sold as a cheap OTC pediatric drug, common in Greece I recall. Some readers of this blog have used it.
I would try a potassium supplement and see if anything good, or bad, happens in the next 15 minutes. Any effect will only be short term.
Thank you so much
ReplyDeleteHello, Peter, this is Grace. Thank you for all these information which helps us to understand Autism better so we can better help our kids. I would like to share this Autism drug news with you: https://scitechdaily.com/groundbreaking-experimental-compound-displays-effectiveness-in-treating-symptoms-of-autism-and-alzheimers-disease/amp/. Sorry if this is not the right post to share that. Do you know more about this experimental drug? Do you think if there is anything similar to it that we can start to try on our kids? Thanks
ReplyDeleteGrace, the short answer is to add wheatgerm to diet, which is proved to increase autophagy, because it contains spermidine. This is also good advice for older relatives. Intranasal ketamine may have a similar effect to what you read about, but unless you have a child with ADNP syndrome, I would not recommend it.
DeleteThe drug in the article you refer to is also known as Davunetide and has long been developed as an intranasal treatment for Alzheimer’s.
Davunetide is the easy name. In the research it also gets called NAP, or more precisely NAPVSIPQ.
Davunetide is derived from a growth factor called activity-dependent neurotrophic protein (ADNP). ADNP contains within it a peptide of the eight amino acids Asn-Ala-Pro-Val-Ser-Ile-Pro-Gln. (NAPVSIPQ).
This peptide has highly potent neuroprotective activity.
One cause of autism with intellectual disability is
In Alzheimer’s there is an accumulation of debris in the brain most likely linked to impaired autophagy, the intracellular garbage collection process. The rather complex science suggests that you can perk up the garbage disposal service and get rid of plaques and tau tangles by treatment with Davunetide (= NAP = NAPVSIPQ).
“The newly discovered ADNP/NAP target FOXO3 controls the autophagy initiator LC3 (microtubule-associated protein 1 light chain 3), with known ADNP binding to LC3 augmented by NAP, protecting against tauopathy. NAP amelioration attests to specificity, with potential for drug development targeting accessible biomarkers. “
Lack of autophagy is a feature of aging and multiple types of dementia.
ADNP syndrome, also known as Helsmoortel-Van Der Aa syndrome, is a neurodevelopmental genetic disorder caused by mutations in the ADNP gene. These mutations occur spontaneously in the majority of reported patients, meaning there has been no family history of the disorder.
The hallmark features of the syndrome are intellectual disability, global developmental delays, global motor planning delays and autism spectrum disorder or autistic features. Although ADNP syndrome was only discovered in 2014, it is projected to be one of the most frequent single gene causes of autism.
Perhaps not surprisingly, in mice given the ADNP mutation there is a lack of ADNP in their brains and this causes an autistic” mouse. The good news is that the impairments are reversed by treating the mouse with the highly active sub-part of ADNP which is NAP = NAPVSIPQ = Davunetide.
Davunetide has been in research from before ADNP syndrome was identified.
If you look up the ADNP gene
https://www.genecards.org/cgi-bin/carddisp.pl?gene=ADNP#:~:text=GeneCards%20Summary%20for%20ADNP%20Gene,this%20gene%20include%20chromatin%20binding.
you will see that it is also associated with agenesis of the corpus callosum, the key bundle of nerves that joins the left and right half of the brain together.
Also listed are autism, ID/MR, Helsmoortel-Van Der Aa Syndrome etc
Ketamine is listed as a possible therapy.
Click in the link and you see that there is a trial:
Low-Dose Ketamine in Children With ADNP Syndrome
DeleteAlexander Kolevzon, Icahn School of Medicine at Mount Sinai
This is a Phase 2A, single dose, open-label study to evaluate the safety, tolerability, and efficacy of a low-dose, 40-minute infusion into the veins (intravenous infusion or "IV") of ketamine in children with ADNP syndrome (Activity-Dependent Neuroprotective Protein). The study team will enroll 10 participants, ages 5 to 12, at Mount Sinai. The study participation is expected to last 4 weeks and will include 5 scheduled clinic visits in order to complete safety monitoring, clinical assessments, and biomarker collection. At the conclusion of this study, the study team expects to demonstrate the safety and tolerability of low-dose ketamine in children with ADNP syndrome. Additionally, the study team anticipates identifying meaningful signals of efficacy in clinical outcome measures using RNA and DNA sequencing to analyze ADNP protein expression and DNA methylation profiles, a natural process by which methyl groups are added to the DNA to change its activity, in order to assess sensitivity to change with low-dose ketamine treatment and inform future phase 3 studies. Ketamine is not currently approved by the Food and Drug Administration to treat this syndrome, but it is approved for use in children in other situations, for example in anesthesia.
Low-dose ketamine drug trial preliminary results are in, and they look very promising.
https://www.adnpfoundation.org/ketamine-update.html
The Seaver Autism Center at the Icahn School of Medicine at Mount Sinai has released preliminary results from the clinical trial of low-dose ketamine, the first ever drug trial for ADNP syndrome.
Note that intranasal Ketamine already is an autism therapy in the research.
Hi Peter, this is Grace. Thank you so much for the information and I appreciate all your help. My daughter has Autism, Epilepsy, Intellectual disability and Anxiety. She does not have ADNP syndrome. She did a genetic testing on 192 genes, the result is inconclusive. The following variants are found: (1)CACNA1A, Exon 1, c.74T>G(p.val25Gly), heterozygous, uncertain significance;
Delete(2)CACNA1H, Exon 20, c.4022C>T(p.Ala1341Val), heterozygous, uncertain significance;
(3)CNTNAP2, Exon 10, c.1636A>G(p.Asn546Asp), heterozygous, uncertain significance;
(4)RYR3, Exon 68, c.9917A>G(p.His3306Arg), heterozygous, uncertain significance;
(5)MICROARRAY CGH Test: Alteration was detected at arr[hg19] CGH Xp22.31(6,488,721-8,097,511)x3.
This duplication has not been reported as a copy number variant from general population.
(6)N-METHYL-D-ASPARTATE RECPT IGG Test: Antibodies to NMDA were not detected. Right now, she has been taking medicine to keep seizure and anxiety under control. She is using the nasal spray Desmopressin Acetate which works wonder for her anxiety. She has tried Bumetanide for several months but has to stop due to side effect of low blood sodium even with salt pill supplements. We did not see much difference from Bumetanide. Peter, for my daughter's case, do you have any suggestion on what to try next especially something that can improve cognitive function? I live in U.S.A. If you know any good doctor that can help with her Autism/Epilepsy/ID, please let me know. Thanks, Grace
Grace, first of all I am very interested that your daughter responds well to Desmopressin nasal spray. This did come up in an earlier post, but in the US doctors seem to usually prescribe specially compounded vasopressin, which is also used in the clinical trials for autism.
DeleteDesmopressin already exists as ready made drug, so would be much more practical. Desmopressin is a modified version of the hormone vasopressin. I would love to know if oral desmopressin gives similar results to intranasal delivery; that would be even simpler.
Which kind of doctor prescribed the Desmopressin?
You have a very interesting list of genes, that look pretty causal to me. I think Dr Chez, the neurologist and autism author would be a good person to talk to. (just google "Dr Chez autism")
You need someone enlightened to prescribe some drugs personalized to the genetic testing results. He apparently is a good guy, but still cautious.
CACNA1A encodes the calcium ion channel Caᵥ2.1, which is a very important one.
You can inhibit CACNA1A with 104 possible substances, mainly common calcium channel blockers, including Verapamil that we use, but also even the diuretic Spironolactone.
https://www.genecards.org/cgi-bin/carddisp.pl?gene=CACNA1A
CACNA1H encodes Voltage-Gated Calcium Channel Subunit Alpha Cav3.2. This ion channel is associated with both epilepsy and autism.
You can inhibit CACNA1H with 73 different possibilities, including an anti seizure drug called Zonisamide, but also with common calcium channel blockers including Verapamil and again spironolactone.
https://www.genecards.org/cgi-bin/carddisp.pl?gene=CACNA1H
It is clearly possible to find a single drug that inhibits both your target calcium channels.
CNTNAP2 is thought of as a master autism gene and is the subject of the above post. You would want to increase EGF somehow.
Apparently EGF production is stimulated by testosterone.
So high estrogen + low testosterone would be a bad thing. I would suggest you check your daughter’s hormone levels.
RYR3 (Ryanodine Receptor 3) is another calcium channel. This time to release calcium from intracellular storage for use in many cellular processes. It does play a role in female hormones. (another reason to check hormones)
It is an epilepsy gene, but it is not seen as an autism gene.
You can modulate it
https://www.genecards.org/cgi-bin/carddisp.pl?gene=RYR3
I think the Desmopressin may have caused the low sodium, when given with bumetanide.
Hello, Peter, this is Grace. Thank you for all the information. The knowledge I got from your posts really bring my understanding of autism to next level. I really appreciate that :). And you are right that my daughter need someone enlightened to prescribe some drugs personalized to the genetic testing results. For the past few years, I have been taking my daughter to see doctors in several big children's hospitals, none of them can explain the cause of her autism/epilepsy/intellectual disability based on all the blood tests and genetic test results and prescribe something helpful. Her brain MRI is normal. I will make an appointment with Dr. Chez. Do you also know other doctor who is specialized in treating autism/epilepsy similar to my daughter's genetic background? The Desmopressin is prescribed by a psychologist. The biggest effect on my daughter is to reduce her sensitivity to sound which in turn greatly improves her anxiety. Now she has no issue to go to crowds or use hand drier or something that could make loud noise. This was mission impossible before Desmopressin. The side effect for Desmopressin is low sodium, so I have to supplement her with salt pills at the same time.
DeleteGrace, in just the 192 genes you analyzed you have several possibly causal genes.
DeleteOne option would be to analyze all 22,000 genes and see what else you find. The problem is that if you cannot treat even the currently identified genes, what is the point.
There is a doctor in California who does the testing, analysis and then tries to apply it. He is called Dr Boles. It is not cheap.
http://molecularmitomd.com/
You could try him as well as Dr Chez.
It is very interesting that Desmopressin is so helpful for sound sensitivity. It may very well be via its effect on electrolytes (sodium down and, in effect, potassium up).
Hi, Peter, this is Grace. Thank you so much. It seems that both Dr. Chez and Dr. Boles are in California. I will try to get appointments with them this summer. Hopefully they will have some enlightening suggestions for my daughter's case. I will come back to update.
DeleteHi Peter, this is Grace. I have been wondering why Bumetanide does not work for my daughter. She is taking a small dosage of Clobazam(10 my per day) together with Oxetellar for her seizure control and responds to them very well. Could that be the reason? Thank you.
DeleteHi Grace, Clobazam is a benzodiazepine class medication, which means it activates the GABAa receptor. In your daughter this produces a calming anti-anxiety/seizure effect. This is a good thing.
DeleteThis also means that in your daughter the important inhibitory neurotransmitter GABA is functioning as nature intended, at least at the GABAa receptor.
In bumetanide responders, GABA functions “in reverse” at GABAa receptors and produces an excitatory effect. In your daughter a movement in the excitatory/inhibitor (E/I) balance towards excitatory would cause seizures. The E/I balance is very important for cognition, but there are multiple possible factors that can disrupt it. Bumetanide partly rectifies just one of these factors.
In practical terms, your daughter does not respond to bumetanide because she does not have the underlying dysfunction that it remedies.
Hi, Peter, this is Grace. Thanks for the explanation. It makes sense. I am still searching for something that could explain her Autism/Epilepsy/ID. I believe they are correlated. So far, I have not been able to find something that really helps her cognitive functions, especially speech. Next I would like to try Choline(Cognizin) to see if that will work for her.
DeleteHi, Peter, this is Grace. I have a quick question. My daughter is not a Bumetanide responder, which means her GABA receptor is functioning. Is this an indicator that she will respond to Choline or not to respond to Choline? Or can we use this information to find something that might help her Autism and cognitive function? Thank you.
DeleteGrace, hundreds of different biological or physical conditions may lead to intellectual disability and/or autism. Some are at least partially treatable and some may not be.
DeleteA very good resource of describing 116 types of treatable ID can be found here:
https://treatable-id.org/
You will notice that some of the treatments are the same, or are clearly not specific to each condition, for example the use of IVIG.
The fact that your daughter is not a bumetanide responder just means you can cross bumetanide off your target list. It does not indicate what to try next.
I think that comorbidities and factors that make your daughter’s symptoms better or worse may give pointers towards what may be effective treatments. Very many people’s autism has an auto-immune component and there are numerous possible therapies for this.
She does have 2 genetic mutations (CACNA1H and CACNA1A) that you can treat very simply with a cheap drug like Verapamil, so you do have some clues to follow up on.
Thank you Peter for the information. Hopefully I can get an appointment with Dr. Chez this summer. I will check Verapimil with him to see if he will prescribe.
DeleteHi Peter, do you have any ideas if anyone has tried skullcap baicalein in autism, it seems that it has some effects associated with suramin, I also found baiclein nasal spray, it is manufactured by a company from Italy, but I don't know if it crosses the blood-brain barrier, forget an article https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0204229
ReplyDeleteDragos, very many people have tried to find an alternative to Suramin, with no success. Suramin is a potent drug and natural substances may share some effects but not to a clinically significant degree.
DeleteIf you want an intra-nasal trial, I would look into Desmopressin/Vasopressin which has been raised several times in this blog. For Grace's daughter the effect is on anxiety, but in other people the effects are broader. There is a lot of research published by Stanford University.
There was a worldwide shortage of Desmopressin intranasal, when I wrote by post about it. It seems to be available again.
HI. ONCE AGAIN I TALK ABOUT MY SON PROBLEM WITH WHITE MATTER. SO I SEE CURCUMIN MAYBE GOOD. BUT BIOVAILABLE IS PROBLEM. WHAT FORM OF CURCUMIN YOU THINK GOOD PETER, LONGVIDA CURCUMIN OR CURCUMIN+PIPERIN SUPPLEMENT ?
ReplyDeleteCurcumin is cheap, but the more bioavailable supplements are expensive. You need large amounts like in Indian food to have a biological effect. Adding black pepper and oil/fat improves absorption. You can make turmeric balls as a cheap bite-sized snack.
Deleteit too hard to my kid eat turmeric. i want buy supplement from iherb or some where but dont know what form of its good
DeleteDear Peter
ReplyDeleteI don't know why I haven't been able to comment on your blog lately (earlier I commented as anonomous, could't make my google account connect).
Anyway my son is 11 and has ASD. We got WGS on him and he has 2 mutations in the CNTNAP2 gene and several others. I am researching as much as I can, but none of his doctors are able to offer advice on possible treatment/supplements. He is on at rather comprensive polypill (thank you for this blog!) and is currently getting:
Quercetin
Luteolin
NAC
Sulphoraphane
Tyrosine
Omega 3/6
L reuteri probiotic
L carnosine
Citicholine
Agmatine
Clemastine
Potassium
And sometimes oxytocin nasal spray
About the genes... I have studies CNTNAP2 - and read your statement about that higner EGF would be good. The testoterone is a little hard to adjust, but what do you think about cow's milk? I read that it contains a good amount of EGF.
His mutations (relevant to autism) are:
CNTNAP2 rs 7794745, allele: TT //and same gene rs2710102, allele: AG)
C9orf72, rs 2814707, allele: CT
SHANK2, rs11237599, allele CT
EN2, rs 1861973, allele: TC, // and same gene rs1861972, allele: GA
MET COMETT, rs 1858830, allele CC
GLO1, rs 4746, allele: TG
He is struggling so much. Fatigue, melt downs, selective mutism because of anxiety, low speed processing, confusion, repetitive behavior. I don't know how to help him better, but I hope that there might be a clever intervention based on the genes...
Also: been thinking about amino acids and piracetam (problem with motor skills). Do you think that could be helpful?
Ps: he cannot use bumetanide, because he suffers from polyuria... Needs to pee constantly. The potassium supplement seems to help somewhat on this. All the bloodwork is "normal", no diabetes or anything like that.
DeleteAm curious to if there are other supplements or medications that could help the way bumetanide does, but without the effect on urination as this is already quite problematic
Piperine should in theory have a similar effect to bumetanide, via a different mechanism - it increases KCC2 expression.
DeleteIt is unkown how much piperine you might need.
I think potassium bromide, the original anti-epilepsy drug from 150 years ago, may lower chloride by replacing it with bromide. Some readers do use it. The drug form for humans is now very expensive, but the basic chemical is very cheap. It is used to treat young children with epilepsy in Europe, but not in the US.
Maria, your son has several mutations and each on their own might cause autism and they are in very different genes. I think you might as well just try and treat "all autism".
DeleteSee if cow's milk is good for him. Try camel's milk. See the effect.
Because of CNTNAP2 I think it is worth a trial of cGPMax. Try 1/2 a capsule a day, see if there is any effect, then try 1 capusle a day.
See if calcium folinate helps. There is a blood test www.fratnow.com
You could try low dose clonazepam for cognition.
If his diet is not good you might find Nystatin helps.
In some cases an SSRI can help.
For myelination you coud try NAG (not NAC) N-Acetyl Glucosamine
Thanks Peter.
DeleteThere is no camel milk to be found in Scandinavia (as far as google says), but will try with more dairy products. He really likes them, yoghurt and milk, but we cut down because he wen't a little low on iron at some point because of it.
Will look into Piperine and all of your suggestions immediately.
You wrote that potassium bromide may lower chloride by replacing it with bromide - I am in no way a scientist so forgive my lack of understanding - but is too hich chloride a typical problem in autism? Would there be any signs/tests to know or is it a case of trial/error to see if it helps on the behaviour and cognition?
It is 'funny' with the genes.... We also got me tested because of other health issues and I have almost the same setup as him and I am not autistic, but have ADD (without the hyperactivity). Never had learning problems and work as a psychologist. Perhaps gender also plays a role.
I read a little about dr. Richard Kelley's work (Evaluation and Treatment of Patients with Autism and Mitochondrial Disease) and his case description matches my son's developmental regression. He was an early talker, extremely alert and was labeled "bright" by others.
He had a tendency to react with high fever on the slightest and developed perioral dermatitis, started having no bladder control after he had been in control of this/not using diapers anymore. And later allergy towards house dust mites. Perhaps it was a combination of these things that set of the regression and a different developmental path.
Anyway, it is a little frustrating to (perhaps) "realise" this now when he is 11, when the regression took place when he was 2,5 years old. I don't know if it even makes sense to try getting testing done at this point.
Some time ago I have bought all of the recommended supplements that dr. Richard Kelley suggests in these cases, but I am a little hesitant. We would have to "stop" the current polypill he is getting, to be able to know what is working, but it also sparks som anxiety and rumination. I am fearing he will deteriorate and it seems as if dr. Richard Kelleys protocol would take some time to show effect - do you have any advice on this dillemma?
Best regards, Maria
One more thing (sorry!): About KCC2. I read that targeting KCC2 "will aid in the development of novel potential therapeutic avenues for strengthening inhibition in ASD and other disorders where an imbalance in excitation/inhibition occurs".
DeleteIs there always an inhibition problem in autistic individuals.
He is physically inactive and seems "hypoactive" besides from discrete stimming, unless he is prompted to move/be active. He has slow processing speed. We got a QEEG (brainmap) and he had very high slow wave activity that should only be there, if one is asleep... And he was awake for the testing.
My totally non scientific way of thinking about this would be that it seems he has too much inhibition.
Maria, here you go for Camel milk. I've used them in the past. They do a good job.
Deletehttps://desertfarms.co.uk/pages/home-delivery
Stephen
Maria, I would just add Dr Kelley's mito cocktail on top of your current therapies. It may mean a lot of pills and powders.
DeleteAs I understand it, his approach is mainly to protect against a further regression. I think a rapid improvement based on his therapy is unlikely. I think l carnitine is the exception, in that in a small group it works wonders quite quickly.
Your son's lack of energy would be consistant with a mitochondrial dysfunction.
The excitatory inhibitory imbalance is sonething different. Circuits in the brain are finely balanced and in autism they are usually out of balance. This usually means neurons firing when they should not and this impairs cognition and may lead to epilepsy in some cases. One type of E/I imbalance is caused by high chloride inside neurons. This is the state the brain is supposed to be at during birth, but there is then supposed to be a developmental switch. In nearly half of severe autism this switch never occurs. Bumetanide is the treatment. The issue is not how much chloride is in the blood, it is how much is inside neurons. There is a test, you just give a benzodiazepine drug like Valium. The effect should calming, but in a bumetanide-responder you will see agitation and even aggression. This is because GABA is acting "in reverse".
I suggest you contact Dr Antonucci in Italy, so that you can access bumetanide and other drug therapies. OTC therapies will not resolve severe types of autism. Many of his patients read this blog. You do not need to take your child to Italy. Just Google "Dr Antonucci autism". Your case has so many relevant mutations I think he would be very interested.
Maria, astaxanthin is also a nkcc1 inhibitor fyi.
Deletehttps://www.ncbi.nlm.nih.gov/pmc/articles/PMC5007682/
Peter, I just cannot thank you enough. Thank you for the guidance and for sparking hope and showing a path forward. I am so touched by it. I will contact Dr Antonucci and see if he would take my son in as a patient. Even if it would require going to Italy, it would be ok - I am just willing to do about anything, if it could help my son further.
DeleteI will try to add the mitochondrial supplements in a couple of days (i am dosing his pills a week in advance). Fortunately he is really good at swallowing pills, when he takes them with a glass of yoghurt - the big ones were a little harder to get down with juice for him,
And thanks to Stephen for the camel milk link and to whom sent the article about astaxanthin. Will definitely look into it.
Best regards, Maria
Good luck and let us know when you find beneficial therapies.
DeleteMy does not seem to respond to NAC -- he is a regressive ASD with high anxiety and sleep issues -- I was reading that NAC can help with autophagic deficiency and decreasing Notch-1/Hes-1 pathway activity. Why would someone not be a responder?
ReplyDeleteFurthermore, astaxanthin also crosses the bbb
ReplyDeletehttps://www.mdpi.com/2227-9059/11/12/3156#:~:text=Astaxanthin%20(AST)%2C%20a%20natural,induced%20ASD%20model%20%5B50%5D.
Peter, do you know anything about stretch injury to astrocytes?
ReplyDeletehttps://scholar.google.com/scholar?hl=en&as_sdt=0%2C23&q=stretch+injury+astrocytes&oq=stretch+injury+astr#d=gs_qabs&t=1716911815889&u=%23p%3DcnZcvlsGPgUJ
Also, have you ever thought of making bumetanide into a liposomal form? Might be able to help it cross the bbb better.
ReplyDeleteStephen
Here is the link. Sorry forgot to add. Nanoparticle bumetanide
ReplyDeletehttps://www.mdpi.com/2079-4991/14/1/113